Preface
Status of This Document
This document presents the specification of the mzTab data format for metabolomics developed by members of the Human Proteome Organisation (HUPO) Proteomics Standards Initiative (PSI) Proteomics Informatics (PI) Working Group, in collaboration with the Metabolomics Standards initiative (MSI). Distribution is unlimited.
Version of This Document
Date created: June 1st, 2012
Last updated: Thu Apr 29 07:08:35 UTC 2021
Based on commit: f52d060232c2de9eb1a2b7f84e5ca48f662433c1 - Commit History
The current version of this document is: version 2.0.0-Final March 2019 (RELEASE)
The latest (draft) version of this document may be found at https://github.com/HUPO-PSI/mzTab.
Type of This Document
This document is a recommendation for a common, community-driven standard data exchange format in metabolomics.
Authors
Please see Chapter 10 for details on the authors and editors of this document.
Abstract
The Metabolomics Standards Initiative (MSI) and the Human Proteome Organisation (HUPO) Proteomics Standards Initiative (PSI) define community standards for data representation in proteomics/metabolomics to facilitate data comparison, exchange and verification. In this context, the two organizations are working together on a shared standard for downstream results, following mass spectrometry (MS) analysis. This document defines a tab-delimited text file format to report metabolomics results, based on a shared core mzTab format, which was primarily used in the proteomics context before (mzTab v 1.0).
The intention of this specification, mzTab for Metabolomics (mzTab-M), is to extend the concepts established in the previous specification, so that more detail can be captured about the evidence trail for quantification, including MS features (different charge states or adducts) and the evidence trail for identifications, specifically for MS-based experiments on small molecules (metabolites, lipids, contaminants, etc.). mzTab-M is not formally backwards compatible, but follows a similar design pattern to simplify adaptation of existing software and to facilitate its integration into bioinformatics processing and submission workflows.
1. Introduction
1.1. Background
This document addresses the systematic description of small molecule identification and quantification data retrieved from mass spectrometry (MS)-based experiments. A large number of software tools are available that analyze MS data and produce a variety of different output data formats.
mzTab-M is intended as a reporting standard for quantitative results from metabolomics/lipodomics approaches. This format is further intended to provide local LIMS systems as well as MS metabolomics repositories a simple way to share and combine basic information.
mzTab has been developed with a view to support the following general tasks (more specific use cases are provided in Chapter 2):
-
Facilitate the sharing of final experimental results, especially with researchers outside the field of metabolomics.
-
Export of results to external software, including programs such as Microsoft Excel® and Open Office Spreadsheet and statistical software / coding languages such as R.
-
Act as an output format of (web-) services that report MS-based results and thus can produce standardized result pages.
-
Be able to link to the external experimental evidence e.g. by referencing back to mzML files.
This document presents a specification, not a tutorial. As such, the presentation of technical details is deliberately direct. The role of the text is to describe the model and justify design decisions made. The document does not discuss how the models should be used in practice, consider tool support for data capture or storage, or provide comprehensive examples of the models in use. It is anticipated that tutorial material will be developed independently of this specification.
1.2. Document Structure
The remainder of this document is structured as follows.
Chapter 2 lists requirements that mzTab-M is designed to support.
Chapter 3 describes the terminology used.
Chapter 4 describes how the specification presented in Chapter 6 relates to other specifications, both those that it extends and those that it is intended to complement.
Chapter 5 discusses the reasoning behind several design decisions taken.
Chapter 6 contains the documentation of the file.
Chapter 7 lists use cases that are currently not supported.
Chapter 8 Conclusions are presented last.
2. Requirements for mzTab
The following requirements have driven the development of the mzTab data model, and are used to define the scope of the format in version 2.0.0.
-
mzTab-M files should be simple enough to make metabolomics results accessible to people outside the respective fields. This should facilitate the sharing of data beyond the borders of the fields and make it accessible to non-experts.
-
mzTab-M files should contain sufficient information to provide an electronic summary of all findings in a metabolomics study to permit its use as a standard documentation format for ‘supplementary material’ sections of publications in metabolomics. It should thus be able to replace PDF tables as a way of reporting small molecules and make published identification and quantification information more accessible.
-
mzTab-M files should enable reporting at different levels of detail: ranging from a simple summary of the final results to a detailed reporting including the experimental design.
-
It should be possible to open mzTab-M files with “standard” software such as Microsoft Excel® or Open Office Spreadsheet. This should furthermore improve the usability of the format to people outside the fields of metabolomics.
-
mzTab-M files should make MS-derived results easily accessible to scripting languages allowing bioinformaticians to develop software without the overhead of developing sophisticated parsing code. Since mzTab-M files will be comparatively small, the data from multiple experiments can be processed at once without requiring special resource management techniques.
-
It should be possible to contain the complete final results of an MS-based metabolomics experiment in a single file, with the exception that different ionisation modes SHOULD be captured in different files (see Section 5.5). This should furthermore reduce the complexity of sharing and processing an experiment’s final results.
-
It should be useful as an output format by web-services that can then be readily accessed by tools supporting mzTab-M.
-
It should be possible to directly link a small molecule record to its source spectrum in an external MS data file.
3. Notational Conventions
The key words “MUST,” “MUST NOT,” “REQUIRED,” “SHALL,” “SHALL NOT,” “SHOULD,” “SHOULD NOT,” “RECOMMENDED,” “MAY,” and “OPTIONAL” are to be interpreted as described in RFC-2119 (Bradner 1997).
4. Relationship to Other Specifications
The specification described in this document has not been developed in isolation; indeed, it is designed to be complementary to, and thus used in conjunction with, several existing and emerging models. Related specifications include the following:
-
mzML (http://www.psidev.info/mzml). mzML is the PSI standard for capturing mass spectra / peak lists resulting from mass spectrometry in proteomics (Martens et al. 2011). mzTab files MAY be used in conjunction with mzML, although it will be possible to use mzTab with other formats of mass spectra. This document does not assume familiarity with mzML.
-
ISA-TAB (http://isa-tools.org/). The ISA framework allows for reporting experimental metadata and study designs in considerable detail, and is already used for describing metabolomics experiments. It is expected that mzTab files may be linked to ISA-TAB formatted files, for cases where a rich experimental design is to be captured. The linkage between mzTab-M and ISA-TAB is further exemplified in section Section 5.12.
4.1. Relationship to mzTab 1.0
The first stable version of mzTab (version 1.0) was developed primarily by the PSI as a format for the final results (identification or quantification) of a proteomics experiment, using MS. In mzTab version 1.0 limited support was included for metabolomics, through a small molecule table, in which end results could be encoded at the level of quantified metabolites. The intention of mzTab-M is to extend these concepts, so that more detail can be captured about the evidence trail for quantification, including MS features (different charge states or adducts) and the evidence trail for identifications - both of which could not be easily supported in mzTab v 1.0. mzTab-M is not formally backwards compatible, but follows a similar design pattern. It has not been designed to support proteomics. However, design decisions made in mzTab-M may in the future be adopted for a version of mzTab specifically intended for proteomics only (mzTab-P). At the time of writing, mzTab version 1.0 remains in active use for proteomics, but is deprecated for use in metabolomics.
4.2. The PSI Mass Spectrometry Controlled Vocabulary (CV)
The PSI-MS controlled vocabulary is intended to provide terms for annotation of mass spectrometry-related file formats. The CV has been generated with a collection of terms from software vendors and academic groups working in the area of mass spectrometry and MS informatics. Some terms describe attributes that must be coupled with a numerical value attribute in the cvParam element (e.g. MS:1000028 “detector resolution”) and optionally a unit for that value (e.g. MS:1001117, “theoretical mass”, units = “dalton”). The terms that require a value are denoted by having a “datatype” key-value pair in the CV itself: MS:1000511 "ms level" value-type:xsd:int. Terms that need to be qualified with units are denoted with a “has_units” key in the CV itself (relationship: has_units: UO:0000221 ! dalton).
As recommended by the PSI CV guidelines, psi-ms.obo should be dynamically maintained via the psidev-ms-vocab@lists.sourceforge.net mailing list that allows any user to request new terms in agreement with the community involved. Once a consensus is reached among the community the new terms are added within a few business days. If there is no obvious consensus, the CV coordinators committee should vote and make a decision. A new psi-ms.obo should then be released by updating the file on the GitHub server without changing the name of the file.
The following ontologies or controlled vocabularies specified below may also be recommended or required in certain instances, as specified within the CV mapping file:
-
Unit Ontology (http://www.obofoundry.org/ontology/uo.html)
-
ChEBI (ftp://ftp.ebi.ac.uk/pub/databases/chebi/ontology/chebi.obo)
-
OBI Ontology of Biological Investigations (http://obi-ontology.org/)
-
NCBITaxon UniProt Taxonomy Database (https://www.ebi.ac.uk/ols/ontologies/ncbitaxon)
-
BRENDA tissue/ enzyme source (http://www.brenda-enzymes.info/ontology/tissue/tree/update/update_files/BrendaTissueOBO).
-
Cell Type ontology (https://raw.githubusercontent.com/obophenotype/cell-ontology/master/cl-basic.obo).
-
Human disease ontology (http://www.disease-ontology.org/).
-
XLMOD ontology with chemical reagents used for cross-linking and derivatization (https://raw.githubusercontent.com/HUPO-PSI/mzIdentML/master/cv/XLMOD.obo).
-
PRIDE (Proteomics Identifications) ontology (https://github.com/PRIDE-Utilities/pride-ontology).
-
MSIO (Metabolomics Standards Initiative) ontology (https://github.com/MSI-Metabolomics-Standards-Initiative/MSIO) for metabolomics, including harvesting, material and sample processing.
5. Resolved Design and scope issues
There were several issues regarding the design of the format that were not clear cut, and a design choice was made that was not completely agreeable to everyone. So that these issues are not continously revisited, we document the issues here and why the decision that is implemented was made.
5.1. Use of identifiers for input spectra to a search
Small molecules MUST be linked to an identifier of the source spectrum (in an external file) from which the identifications are made by way of a reference in the spectra_ref attribute and via the ms_run element which stores the URI of the file in the location attribute.
It is advantageous if there is a consistent system for identifying spectra in different file formats. The following table is implemented in the PSI-MS CV for providing consistent identifiers for different spectrum file formats.
|
Note
|
This table shows examples from the CV but MAY be extended. The CV holds the definite specification for legal encodings of spectrum identifier values. |
| ID | Term | Data type | Comment |
|---|---|---|---|
MS:1000768 |
Thermo nativeID format |
controllerType=xsd:nonNegativeInteger controllerNumber=xsd:positiveInteger scan=xsd:positiveInteger. |
controller=0 is usually the mass spectrometer |
MS:1000769 |
Waters nativeID format |
function=xsd:positiveInteger process=xsd:nonNegativeInteger scan=xsd:nonNegativeInteger |
|
MS:1000770 |
WIFF nativeID format |
sample=xsd:nonNegativeInteger period=xsd:nonNegativeInteger cycle=xsd:nonNegativeInteger experiment=xsd:nonNegativeInteger |
|
MS:1000771 |
Bruker/Agilent YEP nativeID format |
scan=xsd:nonNegativeInteger |
|
MS:1000772 |
Bruker BAF nativeID format |
scan=xsd:nonNegativeInteger |
|
MS:1000773 |
Bruker FID nativeID format |
file=xsd:IDREF |
The nativeID must be the same as the source file ID |
MS:1000774 |
multiple peak list nativeID format |
index=xsd:nonNegativeInteger |
Used for referencing peak list files with multiple spectra, i.e. MGF, PKL, merged DTA files. Index is the spectrum number in the file, starting from 0. |
MS:1000775 |
single peak list nativeID format |
file=xsd:IDREF |
The nativeID must be the same as the source file ID. Used for referencing peak list files with one spectrum per file, typically in a folder of PKL or DTAs, where each sourceFileRef is different |
MS:1000776 |
scan number only nativeID format |
scan=xsd:nonNegativeInteger |
Used for conversion from mzXML, or a DTA folder where native scan numbers can be derived. |
MS:1000777 |
spectrum identifier nativeID format |
spectrum=xsd:nonNegativeInteger |
Used for conversion from mzData. The spectrum id attribute is referenced. |
MS:1001530 |
mzML unique identifier |
xsd:string |
Used for referencing mzML. The value of the spectrum id attribute is referenced directly. |
In mzTab, the spectra_ref attribute should be constructed following the data type specification in CV Terms and Rules. As an example, to reference the third spectrum (index = 2) in an MGF (Mascot Generic Format) file:
MTD ms_run[1]-format [MS, MS:1001062, Mascot MGF file, ] MTD ms_run[1]-id_format [MS, MS:1000774, multiple peak list nativeID format, ] ... SEH ... spectra_ref ... SME ... ms_run[1]:index=2 ...
Example: Reference the spectrum with identifier “scan=11665” in an mzML file.
MTD ms_run[1]-format [MS, MS:1000584, mzML file, ] MTD ms_run[1]-id_format [MS, MS:1001530, mzML unique identifier, ] ... SEH ... spectra_ref ... SME ... ms_run[1]:scan=11665 ...
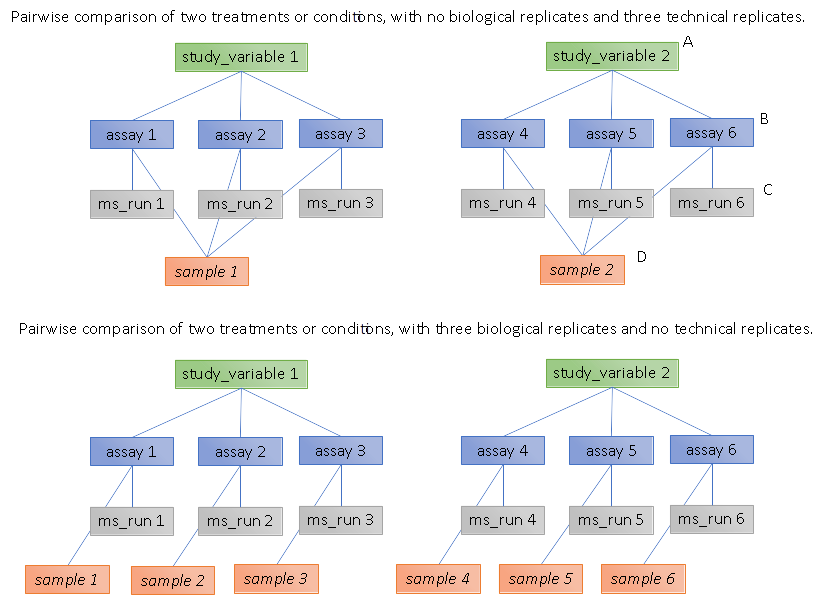
5.2. Recommendations for reporting replicates within experimental designs
Modeling the correct reporting of technical/biological replicates within experimental designs is supported in mzTab as shown in Figure 1. The following components have various cross-references and MUST be used in different types of mzTab files as follows:
-
study_variable – The variables about which the final results of a study are reported, which may have been derived following averaging across a group of replicate measurements (assays). The same concept has been defined by others as “experimental factor”.
-
ms_run – An MS run is effectively one run on an MS instrument, and is referenced from assay in different contexts. In the case of pre-fractionation into n fractions, an assay SHOULD reference n ms_runs.
-
assay – The application of a measurement about the sample (in this case through MS) – producing values about small molecules or lipids. One assay is typically mapped to one MS run in the case of label-free MS analysis (with no pre-fractionation). At the present time, multiplexing within an ms_run is not supported in mzTab-M, thus there would typically be a one:one relationship between assay and ms_run.
-
sample – a biological material that has been analyzed, to which descriptors of species, cell/tissue type etc. can be attached. In all of types of mzTab file, these MAY be reported in the metadata section as sample[1-n]-description. Samples are NOT MANDATORY in mzTab, since many software packages cannot determine what type of sample was analyzed (e.g. whether biological or technical replication was performed). If the file producer wishes to describe whether biological or technical replication has been performed, then sample elements SHOULD be provided.
Clear definitions of biological and technical replicates are difficult to provide as these are somewhat dependent upon the biological domain. However, we use the following general definitions in mzTab.
-
Biological replicates are where different samples have been analyzed by MS.
-
Technical replicates are where same samples are analyzed multiple times by MS.
|
Note
|
There is deliberately no attempt to define the boundary of the term “sample”. |
If sample level information is provided optimally, it is expected that:
-
n biological replicates can be mapped to sample[1-n]
-
m technical replicate measurements of sample 1 SHOULD be mapped to assay[1-m] referencing sample[1] (for example).
However, an open challenge remains since some analysis software is often not aware of whether replicates (multiple MS runs) are originally biological or technical in nature. As such, the default behavior for mzTab exporters from quantitative software is to exclude sample level information and report quantitative data for assay[1-n] and study_variable[1-n].
Additional annotation software would typically be required to add the sample-level information, as provided (often manually) by the user.
5.3. Reporting derivatization approaches
For GC and HPLC, derivatization is often applied in order to specifically target compounds that are otherwise hard to measure at all, being non-volatile or otherwise chemically / physically poorly suited for the separation method and to increase ionization efficiency and selectivity for subsequent MS analysis. For GC, the primary derivatization methods are:
-
acylation
-
alkylation and esterification
-
silylation
In mzTab-M, any derivatization agents used should be reported in the metadata section under derivatization_agent[1-n]. It is expected that in the small molecule evidence table where matches are made to database entries including the derivatized form, then that form SHOULD be reported in evidence row. In the small molecule (summary) table, it MAY be appropriate to reference a database entry for the actual molecule inferred without the derivatization addition, although this is context dependent and in some cases it may be more appropriate to reference a database entry for the derivatized form.
5.4. Encoding missing values, zeroes, nulls, infinity and calculation errors
In the table-based sections there MUST NOT be any empty cells. In case a given property is not available “null” MUST be used, but this is only allowed for parameters with "is nullable=True".
For numerical values, they MUST be encoded following the specifications of xs:decimal. This does not natively support NaN, INF, scientific notation or null. As such, it is allowed in mzTab to include "NaN" for incalculable numbers and "null" for no data. In some cases, there is ambiguity with respect to the use of "0" versus "null": e.g. if there are alignment issues and it is unclear whether a molecule has been quantified with zero abundance or the feature was potentially present in the data but was not found. Export software would be expected to make a decision on this cases, based on best understanding of the case in hand.
Scientific notation and infinity is explicitly not supported.
5.5. Support for positive and negative modes
It is common in metabolomics workflows to use both positive and negative ionisation modes to increase coverage of molecules quantified. In general, an mzTab-M file is intended to capture a data set generated from assays which have been aligned (e.g. in the retention time dimension) to produce a coherent data matrix with few missing values. To our knowledge, it is not common to directly compare the results from positive and negative modes in the same data matrix. As such, we anticipate that such results (i.e. positive mode and negative mode) should be encoded in two different mzTab-M files.
5.6. Referencing evidence for small molecule identifications
Evidence for small molecule identification is captured by reference from the SML table via features (SMFs) down to the final table - Small Molecule Evidence (SME) elements. It is possible to have a legal mzTab-M file that does not contain any features (SML summary level only). In this case, detailed information about small molecule evidence cannot be provided. It is generally RECOMMENDED to include data at the SML, SMF and SME levels.
SMF elements should reference down to all evidence elements (SME rows) that support the identification of that particular feature.
If features (SMF elements) have been grouped prior to evidence collation, then different groups SMF elements SHOULD reference the same SME elements redundantly.
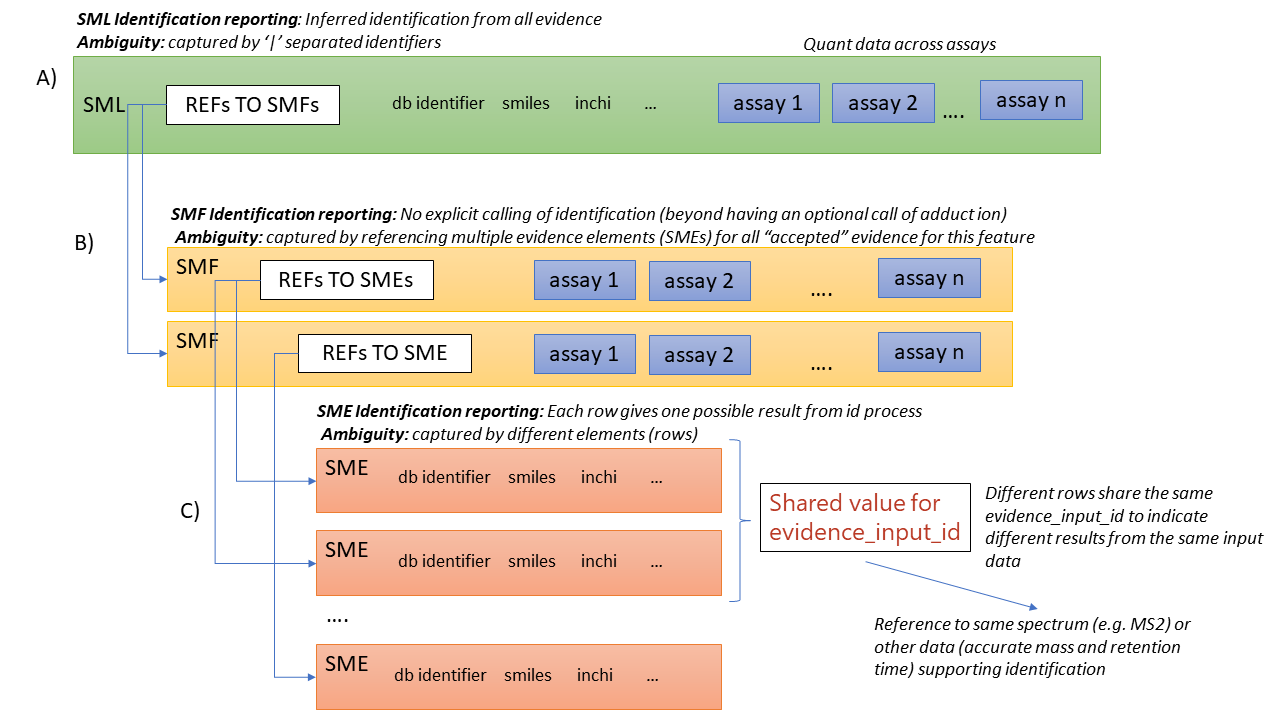
5.7. Ambiguity in identification
It is common in metabolomics and lipidomics for significant ambiguity to remain after data processing in the identification of molecules. In the top level (SML) table, multiple identifiers MAY be provided in several columns: database_identifier, chemical_formula, smiles, inchi, chemical_name and uri. If there is ambiguity in the actual identity of the molecule, multiple identifiers SHOULD be reported separated by the "|" character. The number of elements separated by | characters MUST be identical in all columns where data is reported to emphasize the correspondence across columns.
The SML element Section 6.3.11 MUST be assigned a value to indicate the confidence or ambiguity of the overall assignment. By default, mzTab-M assumes the MSI 4 level system (see Section 6.3.11). A different system of confidence levels MAY be defined in the metadata section (see Section 6.2.57 for details and examples). New systems can be supported in the future by extending the PSI MS controlled vocabulary.
When referencing from the features (SMF) elements to evidence (SME) elements, it is possible for a SMF element to reference multiple SME elements. However, there are potentially several reasons for a 1 to many relationship. A different code MUST be provided in the SME_ID_REF_ambiguity_code element to clarify the case:
-
The same input data (e.g. fragment spectrum or isotopic profile) has multiple results, supporting different potential identifications i.e. where ambiguity remains (code=1)
-
Different input data (or different searches of the same data) have returned results evidence supporting the same identification i.e. no ambiguity remains (code=2).
-
Different input data has been used to support identification and ambiguity still remains (code=3).
5.8. Ambiguity in lipidomics identification
The mzTab-M 2.0.0 release is intended to be used for capturing profiling studies from both metabolomics and lipidomics. However, it is acknowledged that representing ambiguity in the identification of lipid molecules, based on the available evidence from MS is potentially more complicated than for small molecules. As such, mzTab-M 2.0.0 SHOULD be used on release for representing lipid-based data, but a working group will continue to improve on the mechanism for representing lipid identification data, for example defining particular CV terms to be used in the appropriate places of the standard. These artefacts will be reported in due course and should plug-in to this version in a backwards-compatible manner.
5.9. Guidelines for reporting results prior to or with no alignment step across features
The most common intended use for mzTab-M is to encode MS results that have been aligned across multiple analyses (assays), for example by retention time alignment in LC-MS or GC-MS approaches. However, it is possible to use mzTab-M as part of internal pipelines to represent small molecules quantified by MS (features) before alignment. The RECOMMENDED encoding for doing this would be to represent the features from n MS analyes in n mzTab files, rather than attempting to create an SMF table including a sparse matrix filled with nulls for all but one of the assay columns.
5.10. Guidelines for workflows involving pre-fractionation
It is possible that a single analysis of a sample is split offline via some fractionation technology prior to LC/GC-MS into n MS analyses to limit the complexity of the molecules arriving at the detector. Such workflows, while relatively rare in metabolomics, can be encoded in mzTab-M via an assay referencing to n ms_runs. It may be desirable to maintain the link from a feature (SMF row) to the ms_run from which it was obtained. This SHOULD be achieved through the use of an optional column called "opt_global_ms_run_refs", in which the identifiers of ms_runs are placed where the feature has been quantified from.
5.11. Adding optional columns
Additional columns MAY be added to the end of rows in all the table-based sections. The information stored within an optional column is completely up to the resource that generates the file. It MUST not be assumed that optional columns having the same name in different mzTab files contain the same type of information.
These column headers MUST start with the prefix “opt_” followed by the identifier of the object they reference: assay, study variable, MS run or “global” (if the value relates to all replicates). Column names MUST only contain the following characters: ‘A’-‘Z’, ‘a’-‘z’, ‘0’-‘9’, ‘’, ‘-’, ‘[’, ‘]’, and ‘:’. CV parameter accessions MAY be used for optional columns following the format: opt_{OBJECT_ID}_cv_{accession}_{parameter name}. Spaces within the parameter’s name MUST be replaced by ‘’.
COM Example showing a global aligned 2D feature retention time for GCxGC-MS … SFH SMF_ID … opt_global_retention_time_nd SMF 1 … 1562 | 2.47
COM Example showing how drift time values are reported in an additional column from MS run 1 using COM MS CV parameter “ion mobility drift time” (MS:1002476) … SFH SMF_ID … opt_ms_run[1]_cv_MS:MS:1002476_ion_mobility_drift_time SMF 1 … 24.55
5.12. Referencing external resources
The ISA-TAB format (Sansone 2012) is designed for capturing rich experimental designs in terms of a workflow of protocol steps, covering sample processing, data collection and data analysis for any type of high-throughput study. mzTab-M does not aim for a rich description of protocols, but is instead focused on tightly defining the data output from a metabolomics study. Users may wish to use ISA-TAB to record more details about these aspects. The ISA-TAB file can be referenced by the external_study_uri attribute.
Generally, any external resource reference (suffixed -uri, or -location) must be provided as a valid URI string. This allows to report local, as well as remote resource links (URLs) and unique unified resource names (URNs).
Reporting database identifiers SHOULD be kept compatible to http://identifiers.org/, as is demonstrated in the Section 6.3.3 examples, where the database identifier must be preceded by the resource description (prefix) followed by a colon, as specified in the Section 6.2.50 metadata section. The possible use of the full identifiers.org URI is shown in the example for uri attribute within the SML section (Section 6.3.8).
5.13. Other supporting materials
Example files are located at GitHub.
6. Format specification
This section describes the structure of an mzTab file.
-
Field separator
The column delimiter is the Unicode Horizontal Tab character (Unicode codepoint 0009). -
File encoding
The UTF-8 encoding of the Unicode character set is the preferred encoding for mzTab files. However, parsers should be able to recognize commonly used encodings. -
Case sensitivity
All column labels and field names are case-sensitive. -
Line prefix
Every line in an mzTab file MUST start with a three letter code identifying the type of line delimited by a Tab character. The three letter codes are as follows:-
MTDfor metadata -
SMHfor small molecule table header line (the column labels) -
SMLfor rows of the small molecule table -
SFHfor small molecule feature header line -
SMFfor rows of the small molecule feature table -
SEHfor small molecule evidence header line -
SMEfor rows of the small molecule evidence table -
COMfor comment lines
-
-
Header lines
Each table based section (small molecule, small molecule feature and small molecule evidence) MUST start with the corresponding header line. These header lines MUST only occur once in the document since each section also MUST only occur once. -
Dates
Dates and times MUST be supplied in the ISO 8601 format (“YYYY-MM-DD”, “YYYY-MM-DDTHH:MMZ” respectively). -
Decimal separator
In mzTab files the dot (“.”) MUST be used as decimal separator. Thousand separators MUST NOT be used in mzTab files. -
Comment lines and empty lines
Comment lines can be placed anywhere in an mzTab file. These lines must start with the three-letter code COM and are ignored by most parsers. Empty lines can also occur anywhere in an mzTab file and are ignored. -
Params
mzTab makes use of CV parameters. As mzTab is expected to be used in several experimental environments where parameters might not yet be available for the generated scores etc. all parameters can either report CV parameters or user parameters that only contain a name and a value.
Parameters are always reported as[CV label, accession, name, value]. Any field that is not available MUST be left empty.
[MS, MS:1001477, SpectraST,] [,,A user parameter, The value]
Should the name of the param contain commas, quotes MUST be added to avoid problems with the parsing: [label, accession, “first part of the param name, second part of the name”, value].
[MOD, MOD:00648, "N,O-diacetylated L-serine",]
A CV parameter mapping file for mzTab following the mzML mapping file XML Schema is available at GitHub as part of the specification for semantic validation. The mapping file defines recommended controlled vocabularies and defines restrictions for the use of CV terms on particular elements of the mzTab document. Unlike other PSI standards, the model description of mzTab-M 2.0 is not based on an XML schema, but instead on a Swagger / OpenAPI 2.0 compatible specification that is used to generate a corresponding object structure that can be represented in XML, JSON or as an object hierarchy in arbitrary programming languages.
-
Sample IDs
To be able to supply metadata specific to each sample, ids in the formatsample[1-n]are used.
MTD sample[1]-species[1] [NCBITaxon, NCBITaxon:9606, Homo sapiens, ]
-
Assay IDs
To be able to supply metadata specific to each assay, ids in the formatassay[1-n]are used.
MTD assay[1] first assay description
-
Study variable IDs
To be able to supply metadata specific to each study variable (grouping of assays), ids in the formatstudy_variable[1-n]are used.
MTD study_variable[1] Group B (spike-in 0.74 fmol/uL)
-
URIs
URIs MUST follow the format defined in RFC 3986 and RFC 8089 ('file' URIs). -
Versioning
To support a future evolution of the format, an mzTab file MUST report its version. From version 2.0.0-M onwards, we intend to use semantic versioning. This means that increasing the last digit of the version (the patch level) indicates backwards compatible fixes to the specification that require no adaptation of consumers or producers of the format. A change in the middle digit of the version (the minor level) indicates new features that are backwards compatible to existing software but will require updates for new producers and consumers to make use of those features. Finally, a change in the first digit of the version (the major level) indicates breaking changes in the format that require changes in any producing or consuming software to support features of that version.
6.1. Sections
The mzTab-M format consists of four cross-referenced data tables (Figure 3): metadata (MTD), Small Molecule (SML), Small Molecule Feature (SMF) and the Small Molecule Evidence (SME). The MTD and SML tables are mandatory, and for a file to contain any evidence about how molecules were quantified or identified by software, then all four tables must be present. The tables must follow the order MTD, SML, SMF and SME, with a blank line separating each table. The structure of each table, in terms of the rows and columns that must be present is tightly specified, as explained in the following sections, and formally in the mzTab-M specification document.
mzTab-M files MUST have one Metadata (MTD) section and one Small Molecule (SML) Section. In practice, we expect that most files SHOULD also include one Small Molecule Feature (SMF) section, and one Small Molecule Evidence (SME) Section. Files lacking SMF and SME sections can only present summary data about quantified molecules, without any evidence trail for how those values were derived. It will be left to reading software to determine whether additional validation will be requested such that SMF and SME tables MUST be present.

6.2. Metadata Section
The metadata section provides additional information about the dataset(s) reported in the mzTab file. All fields in the metadata section are optional apart from those noted as mandatory. The fields in the metadata section MUST be reported in order of the various fields listed here. The field’s name and value MUST be separated by a tab character:
MTD publication [MS, MS:1000879, PubMed identifier, 12345]
In the following list of fields any term encapsulated by {} is meant as a variable which MUST be replaced accordingly.
Regular expressions (Regex) follow the Perl regular expression syntax with minimal escaping.
Core Metadata
6.2.1. mzTab-version
Description |
The version of the mzTab file. The suffix MUST be "-M" for mzTab for metabolomics (mzTab-M). |
Type |
Regex \d{2}\.\d{0}\.\d{0}-M
|
Mandatory |
True |
Example |
MTD mzTab-version 2.0.0-M |
6.2.2. mzTab-ID
Description |
The ID of the mzTab file, this could be supplied by the repository from which it is downloaded or a local identifier from the lab producing the file. It is not intended to be a globally unique ID but carry some locally useful meaning. |
Type |
String |
Mandatory |
True |
Example |
MTD mzTab-ID MTBL1234 |
6.2.3. title
Description |
The file’s human readable title. |
Type |
String |
Mandatory |
False |
Example |
MTD title Effects of Rapamycin on metabolite profile |
6.2.4. description
Description |
The file’s human readable description. |
Type |
String |
Mandatory |
False |
Example |
MTD description An experiment investigating the effects of Il-6. |
6.2.5. sample_processing[1-n]
Description |
A list of parameters describing a sample processing, preparation or handling step similar to a biological or analytical methods report. The order of the sample_processing items should reflect the order these processing steps were performed in. If multiple parameters are given for a step these MUST be separated by a “|”. If derivatization was performed, it MUST be reported here as a general step, e.g. 'silylation' and the actual derivatization agens MUST be specified in the Section 6.2.54 part. |
Type |
Parameter List |
Mandatory |
False |
Example |
MTD sample_processing[1] [MSIO, MSIO:0000107, metabolism quenching using precooled 60 percent methanol ammonium bicarbonate buffer,] MTD sample_processing[2] [MSIO, MSIO:0000146, centrifugation,] MTD sample_processing[3] [MSIO, MSIO:0000141, metabolite extraction,] MTD sample_processing[4] [MSIO, MSIO:0000141, silylation,] |
6.2.6. instrument[1-n]-name
Description |
The name of the instrument used in the experiment. Multiple instruments are numbered 1..n. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD instrument[1]-name [MS, MS:1000449, LTQ Orbitrap,] |
6.2.7. instrument[1-n]-source
Description |
The instrument’s source used in the experiment. Multiple instruments are numbered [1-n]. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD instrument[1]-source [MS, MS:1000073, ESI,] … MTD instrument[2]-source [MS, MS:1000598, ETD,] |
6.2.8. instrument[1-n]-analyzer[1-n]
Description |
The instrument’s analyzer type used in the experiment. Multiple instruments are numbered [1-n]. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD instrument[1]-analyzer[1] [MS, MS:1000291, linear ion trap,] … MTD instrument[2]-analyzer[1] [MS, MS:1000484, orbitrap,] |
6.2.9. instrument[1-n]-detector
Description |
The instrument’s detector type used in the experiment. Multiple instruments are numbered [1-n]. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD instrument[1]-detector [MS, MS:1000253, electron multiplier,] … MTD instrument[2]-detector [MS, MS:1000348, focal plane collector,] |
6.2.10. software[1-n]
Description |
Software used to analyze the data and obtain the reported results. The parameter’s value SHOULD contain the software’s version. The order (numbering) should reflect the order in which the tools were used. |
Type |
Parameter |
Mandatory |
True |
Example |
MTD software[1] [MS, MS:1002879, Progenesis QI, 3.0] |
6.2.11. software[1-n]-setting[1-n]
Description |
A software setting used. This field MAY occur multiple times for a single software. The value of this field is deliberately set as a String, since there currently do not exist CV terms for every possible setting. |
Type |
String |
Mandatory |
False |
Example |
MTD software[1]-setting Fragment tolerance = 0.1 Da … MTD software[2]-setting Parent tolerance = 0.5 Da |
6.2.12. publication[1-n]
Description |
A publication associated with this file. Several publications can be given by indicating the number in the square brackets after “publication”. PubMed ids must be prefixed by “pubmed:”, DOIs by “doi:”. Multiple identifiers MUST be separated by “|”. |
Type |
String |
Mandatory |
False |
Example |
MTD publication[1] pubmed:21063943|doi:10.1007/978-1-60761-987-1_6 MTD publication[2] pubmed:20615486|doi:10.1016/j.jprot.2010.06.008 |
6.2.13. contact[1-n]-name
Description |
The contact’s name. Several contacts can be given by indicating the number in the square brackets after "contact". A contact has to be supplied in the format [first name] [initials] [last name] (see example). |
Type |
String |
Mandatory |
False |
Example |
MTD contact[1]-name James D. Watson … MTD contact[2]-name Francis Crick |
6.2.14. contact[1-n]-affiliation
Description |
The contact’s affiliation. |
Type |
String |
Mandatory |
False |
Example |
MTD contact[1]-affiliation Cambridge University, UK MTD contact[2]-affiliation Cambridge University, UK |
6.2.15. contact[1-n]-email
Description |
The contact’s e-mail address. |
Type |
String |
Mandatory |
False |
Example |
MTD contact[1]-email watson@cam.ac.uk … MTD contact[2]-email crick@cam.ac.uk |
6.2.16. uri[1-n]
Description |
A URI pointing to the file’s source data (e.g., a MetaboLights records). |
Type |
URI |
Mandatory |
False |
Example |
MTD uri[1] https://www.ebi.ac.uk/metabolights/MTBLS517 |
6.2.17. external_study_uri[1-n]
Description |
A URI pointing to an external file with more details about the study design (e.g., an ISA-TAB file). |
Type |
URI |
Mandatory |
False |
Example |
MTD external_study_uri[1] https://www.ebi.ac.uk/metabolights/MTBLS517/files/i_Investigation.txt |
6.2.18. quantification_method
Description |
The quantification method used in the experiment reported in the file. |
Type |
Parameter |
Mandatory |
True |
Example |
MTD quantification_method [MS, MS:1001834, LC-MS label-free quantitation analysis, ] MTD quantification_method [MS, MS:1001838, SRM quantitation analysis, ] |
6.2.19. sample[1-n]
Description |
A name for each sample to serve as a list of the samples that MUST be reported in the following tables. Samples MUST be reported if a statistical design is being captured (i.e. bio or tech replicates). If the type of replicates are not known, samples SHOULD NOT be reported. |
Type |
String |
Mandatory |
False |
Example |
MTD sample[1] individual number 1 MTD sample[2] individual number 2 |
6.2.20. sample[1-n]-species[1-n]
Description |
The respective species of the samples analysed. For more complex cases, such as metagenomics, optional columns and userParams should be used. |
Type |
Parameter |
Mandatory |
False |
Example |
COM Experiment where all samples consisted of the same two species MTD sample[1]-species[1] [NCBITaxon, NCBITaxon:9606, Homo sapiens, ] MTD sample[2]-species[1] [NCBITaxon, NCBITaxon:39767, Human rhinovirus 11, ] COM Experiment where two samples from different species (combinations) COM were analysed as biological replicates. MTD sample[1]-species[1] [NCBITaxon, NCBITaxon:9606, Homo sapiens, ] MTD sample[1]-species[2] [NCBITaxon, NCBITaxon:39767, Human rhinovirus 11, ] MTD sample[2]-species[1] [NCBITaxon, NCBITaxon:9606, Homo sapiens, ] MTD sample[2]-species[2] [NCBITaxon, NCBITaxon:12130, Human rhinovirus 2, ] |
6.2.21. sample[1-n]-tissue[1-n]
Description |
The respective tissue(s) of the sample. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD sample[1]-tissue[1] [BTO, BTO:0000759, liver, ] |
6.2.22. sample[1-n]-cell_type[1-n]
Description |
The respective cell type(s) of the sample. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD sample[1]-cell_type[1] [CL, CL:0000182, hepatocyte, ] |
6.2.23. sample[1-n]-disease[1-n]
Description |
The respective disease(s) of the sample. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD sample[1]-disease[1] [DOID, DOID:684, hepatocellular carcinoma, ] MTD sample[1]-disease[2] [DOID, DOID:9451, alcoholic fatty liver, ] |
6.2.24. sample[1-n]-description
Description |
A human readable description of the sample. |
Type |
String |
Mandatory |
False |
Example |
MTD sample[1]-description Hepatocellular carcinoma samples. MTD sample[2]-description Healthy control samples. |
6.2.25. sample[1-n]-custom[1-n]
Description: |
Parameters describing the sample’s additional properties. Dates MUST be provided in ISO-8601 format. |
Type: |
Parameter |
Mandatory |
False |
Example |
MTD sample[1]-custom[1] [,,Extraction date, 2011-12-21] MTD sample[1]-custom[2] [,,Extraction reason, liver biopsy] |
6.2.26. ms_run[1-n]-location
Description |
Location of the external data file e.g. raw files on which analysis has been performed. If the actual location of the MS run is unknown, a “null” MUST be used as a place holder value, since the [1-n] cardinality is referenced elsewhere. If pre-fractionation has been performed, then [1-n] ms_runs SHOULD be created per assay. |
Type |
URI |
Mandatory |
True |
Example |
MTD ms_run[1]-location file:///C:/path/to/my/file … MTD ms_run[1]-location ftp://ftp.ebi.ac.uk/path/to/file |
6.2.27. ms_run[1-n]-instrument_ref
Description |
If different instruments are used in different runs, this attribute can be used to link a specific instrument to a specific run. |
Type |
Integer |
Mandatory |
False |
Example |
MTD ms_run[1]-instrument_ref instrument[1] |
6.2.28. ms_run[1-n]-format
Description |
A parameter specifying the data format of the external MS data file. If ms_run[1-n]-format is present, ms_run[1-n]-id_format SHOULD also be present, following the parameters specified in Table 1. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD ms_run[1]-format [MS, MS:1000584, mzML file, ] MTD ms_run[1]-id_format [MS, MS:1000530, mzML unique identifier, ] … MTD ms_run[2]-format [MS, MS:1001062, Mascot MGF file, ] MTD ms_run[2]-id_format [MS, MS:1000774, multiple peak list nativeID format, ] |
6.2.29. ms_run[1-n]-id_format
Description |
Parameter specifying the id format used in the external data file. If ms_run[1-n]-id_format is present, ms_run[1-n]-format SHOULD also be present. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD ms_run[1]-format [MS, MS:1000584, mzML file, ] MTD ms_run[1]-id_format [MS, MS:1000530, mzML unique identifier, ] … MTD ms_run[2]-format [MS, MS:1001062, Mascot MGF file, ] MTD ms_run[2]-id_format [MS, MS:1000774, multiple peak list nativeID format, ] |
6.2.30. ms_run[1-n]-fragmentation_method[1-n]
Description |
The type(s) of fragmentation used in a given ms run. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD ms_run[1]-fragmentation_method[1] [MS, MS:1000133, CID, ] … MTD ms_run[1]-fragmentation_method[2] [MS, MS:1000422, HCD, ] |
6.2.31. ms_run[1-n]-scan_polarity[1-n]
Description |
The polarity mode of a given run. Usually only one value SHOULD be given here except for the case of mixed polarity runs. |
Type |
Parameter |
Mandatory |
True |
Example |
MTD ms_run[1]-scan_polarity[1] [MS, MS:1000130, positive scan, ] OR MTD ms_run[1]-scan_polarity[1] [MS, MS:1000129, negative scan, ] OR (For mixed polarity in one run) MTD ms_run[1]-scan_polarity[1] [MS, MS:1000130, positive scan, ] MTD ms_run[1]-scan_polarity[2] [MS, MS:1000129, negative scan, ] |
6.2.32. ms_run[1-n]-hash
Description |
Hash value of the corresponding external MS data file defined in ms_run[1-n]-location. If ms_run[1-n]-hash is present, ms_run[1-n]-hash_method SHOULD also be present. |
Type |
String |
Mandatory |
False |
Example |
MTD ms_run[1]-hash_method [MS, MS:1000569, SHA-1, ] MTD ms_run[1]-hash de9f2c7fd25e1b3afad3e85a0bd17d9b100db4b3 |
6.2.33. ms_run[1-n]-hash_method
Description |
A parameter specifying the hash methods used to generate the String in ms_run[1-n]-hash. Specifics of the hash method used MAY follow the definitions of the mzML format. If ms_run[1-n]-hash is present, ms_run[1-n]-hash_method SHOULD also be present. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD ms_run[1]-hash_method [MS, MS:1000569, SHA-1, ] MTD ms_run[1]-hash de9f2c7fd25e1b3afad3e85a0bd17d9b100db4b3 |
6.2.34. assay[1-n]
Description |
A name for each assay, to serve as a list of the assays that MUST be reported in the following tables. |
Type |
String |
Mandatory |
True |
Example |
MTD assay[1] first assay MTD assay[2] second assay |
6.2.35. assay[1-n]-custom[1-n]
Description |
Additional parameters or values for a given assay. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD assay[1]-custom[1] [MS, , Assay operator, Fred Blogs] |
6.2.36. assay[1-n]-external_uri
Description |
A reference to further information about the assay, for example via a reference to an object within an ISA-TAB file. |
Type |
URI |
Mandatory |
False |
Example |
MTD assay[1]-external_uri https://www.ebi.ac.uk/metabolights/MTBLS517/files/i_Investigation.txt?STUDYASSAY=a_e04_c18pos.txt |
6.2.37. assay[1-n]-sample_ref
Description |
An association from a given assay to the sample analysed. |
Type |
{SAMPLE_ID} |
Mandatory |
False |
Example |
MTD assay[1]-sample_ref sample[1] MTD assay[2]-sample_ref sample[2] |
6.2.38. assay[1-n]-ms_run_ref
Description |
An association from a given assay to the source MS run. All assays MUST reference exactly one ms_run unless a workflow with pre-fractionation is being encoded, in which case each assay MUST reference n ms_runs where n fractions have been collected. Multiple assays SHOULD reference the same ms_run to capture multiplexed experimental designs. |
Type |
{MS_RUN_ID} |
Mandatory |
True |
Example |
MTD assay[1]-ms_run_ref ms_run[1] MTD assay[1]-ms_run_ref ms_run[2] |
6.2.39. study_variable[1-n]
Description |
A name for each study variable (experimental condition or factor), to serve as a list of the study variables that MUST be reported in the following tables. For software that does not capture study variables, a single study variable MUST be reported, linking to all assays. This single study variable MUST have the identifier “undefined“. |
Type |
String |
Mandatory |
True |
Example |
MTD study_variable[1] “control” MTD study_variable[2] “1 minute” |
6.2.40. study_variable[1-n]-assay_refs
Description |
Bar-separated references to the IDs of assays grouped in the study variable. |
Type |
{ASSAY_ID}, … |
Mandatory |
True |
Example |
MTD study_variable[1]-assay_refs assay[1]| assay[2]| assay[3] |
6.2.41. study_variable[1-n]-average_function
Description |
The function used to calculate the study variable quantification value and the operation used is not arithmetic mean (default) e.g. “geometric mean”, “median”. The 1-n refers to different study variables. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD study_variable-average_function [MS, MS:1002883, median, ] |
6.2.42. study_variable[1-n]-variation_function
Description |
The function used to calculate the study variable quantification variation value if it is reported and the operation used is not coefficient of variation (default) e.g. “standard error”. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD study_variable-variation_function [MS, MS:1002885, standard error, ] |
6.2.43. study_variable[1-n]-description
Description |
A textual description of the study variable. |
Type |
String |
Mandatory |
True |
Example |
MTD study_variable[1]-description Group B (spike-in 0.74 fmol/uL) |
6.2.44. study_variable[1-n]-factors
Description |
Additional parameters or factors, separated by bars, that are known about study variables allowing the capture of more complex, such as nested designs. |
Type |
Param List |
Mandatory |
False |
Example |
MTD study_variable[1]-factors [,,rapamycin dose,0.5mg] |
6.2.45. custom[1-n]
Description |
Any additional parameters describing the analysis reported. |
Type |
Parameter |
Mandatory |
False |
Example |
MTD custom[1] [,,MS operator, Florian] |
6.2.46. cv[1-n]-label
Description |
A string describing the labels of the controlled vocabularies/ontologies used in the mzTab file as a short-hand e.g. "MS" for PSI-MS. |
Type |
String |
Mandatory |
True |
Example |
MTD cv[1]-label MS |
6.2.47. cv[1-n]-full_name
Description |
A string describing the full names of the controlled vocabularies/ontologies used in the mzTab file. |
Type |
String |
Mandatory |
True |
Example |
MTD cv[1]-full_name PSI-MS controlled vocabulary |
6.2.48. cv[1-n]-version
Description |
A string describing the version of the controlled vocabularies/ontologies used in the mzTab file. |
Type |
String |
Mandatory |
True |
Example |
MTD cv[1]-version 4.1.11 |
6.2.49. cv[1-n]-uri
Description |
A string containing the URIs of the controlled vocabularies/ontologies used in the mzTab file. |
Type |
String |
Mandatory |
True |
Example |
MTD cv[1]-uri https://raw.githubusercontent.com/HUPO-PSI/psi-ms-CV/master/psi-ms.obo |
6.2.50. database[1-n]
Description |
The description of databases used. For cases, where a known database has not been used for identification, a userParam SHOULD be inserted to describe any identification performed e.g. de novo. If no identification has been performed at all then "no database" should be inserted followed by null. |
Type |
Param |
Mandatory |
True |
Example |
MTD database[1] [MIRIAM, MIR:00100079, HMDB, ] MTD database[2] [,, "de novo", ] MTD database[3] [MIRIAM, MIR:00000002, CHEBI, ] MTD database[4] [,, "customDB", ] OR MTD database[5] [,, "no database", null ] |
6.2.51. database[1-n]-prefix
Description |
The prefix used in the “identifier” column of data tables. For the “no database” case "null" must be used. |
Type |
String |
Mandatory |
True |
Example |
MTD database[1]-prefix hmdb MTD database[2]-prefix dn MTD database[3]-prefix chebi MTD database[4]-prefix cust OR MTD database[5]-prefix null |
6.2.52. database[1-n]-version
Description: |
The database version is mandatory where identification has been performed. This may be a formal version number e.g. “1.4.1”, a date of access “2016-10-27” (ISO-8601 format) or “Unknown” if there is no suitable version that can be annotated. |
Type: |
String |
Mandatory |
True |
Example |
MTD database[1]-version 3.6 OR MTD database[2]-version Unknown |
6.2.53. database[1-n]-uri
Description |
The URI to the database. For the “no database” case, "null" must be reported. |
Type |
URI |
Mandatory |
True |
Example |
MTD database[1]-uri http://www.hmdb.ca/ OR MTD database[5]-uri null |
6.2.54. derivatization_agent[1-n]
Description |
A description of derivatization agents applied to small molecules, using userParams or CV terms where possible. |
Type |
Param |
Mandatory |
False |
Example |
MTD derivatization_agent[1] [XLMOD, XLMOD:07014, N-methyl-N-t-butyldimethylsilyltrifluoroacetamide, ] |
6.2.55. small_molecule-quantification_unit
Description |
Defines what type of units are reported in the small molecule summary quantification / abundance fields. |
Type |
Parameter |
Mandatory |
True |
Example |
MTD small_molecule-quantification_unit [MS, MS:1002887, Progenesis QI normalised abundance, ] |
6.2.56. small_molecule_feature-quantification_unit
Description |
Defines what type of units are reported in the small molecule feature quantification / abundance fields. |
Type |
Parameter |
Mandatory |
True (if SMF section is being reported) |
Example |
MTD small_molecule_feature-quantification_unit [MS, MS:1002887, Progenesis QI normalised abundance, ] |
6.2.57. small_molecule-identification_reliability
Description |
The system used for giving reliability / confidence codes to small molecule identifications MUST be specified if not using the default codes (see Section 6.3.11 and for details). |
Type |
Param |
Mandatory |
False |
Example |
MTD small_molecule-identification_reliability [MS, MS:1002896, compound identification confidence level, ] or MTD small_molecule-identification_reliability [MS, MS:1002955, hr-ms compound identification confidence level, ] |
6.2.58. id_confidence_measure[1-n]
Description |
The type of small molecule confidence measures or scores MUST be reported as a CV parameter [1-n]. The CV parameter definition should formally state whether the ordering is high to low or vice versa. The order of the scores SHOULD reflect their importance for the identification and be used to determine the identification’s rank. |
Type |
Parameter |
Mandatory |
True |
Example |
id_confidence_measure[1] [MS,MS:1002889,Progenesis MetaScope Score,] id_confidence_measure[2] [MS,MS:1002890,fragmentation score,] id_confidence_measure[3] [MS,MS:1002891,isotopic fit score,] |
6.2.59. colunit-small_molecule
Description |
Defines the used unit for a column in the small molecule section. The format of the value has to be \{column name}=\{Parameter defining the unit} This field MUST NOT be used to define a unit for quantification columns. The unit used for small molecule quantification values MUST be set in small_molecule-quantification_unit. |
Type |
String |
Mandatory |
False |
Example |
MTD colunit-small_molecule opt_global_cv_MS:MS:1002954_collisional_cross_sectional_area=[UO,UO:00003241, square angstrom,] |
6.2.60. colunit-small_molecule_feature
Description |
Defines the used unit for a column in the small molecule feature section. The format of the value has to be \{column name}=\{Parameter defining the unit} This field MUST NOT be used to define a unit for quantification columns. The unit used for small molecule quantification values MUST be set in small_molecule_feature-quantification_unit. |
Type |
String |
Mandatory |
False |
Example |
MTD colunit-small_molecule_feature opt_ms_run[1]_cv_MS:MS:1002476_ion_mobility_drift_time=[UO,UO:0000031, minute,] |
6.2.61. colunit-small_molecule_evidence
Description |
Defines the used unit for a column in the small molecule evidence section. The format of the value has to be \{column name}=\{Parameter defining the unit}. |
Type |
String |
Mandatory |
False |
Example |
MTD colunit-small_molecule_evidence opt_global_mass_error=[UO, UO:0000169, parts per million, ] |
6.3. Small Molecule Section
The small molecule section is table-based. The small molecule section MUST always come after the metadata section. All table columns MUST be Tab separated. There MUST NOT be any empty cells; missing values MUST be reported using “null” for columns where Is Nullable = “True”.
Each row of the small molecule section is intended to report one final result to be communicated in terms of a molecule that has been quantified. In many cases, this may be the molecule of biological interest, although in some cases, the final result could be a derivatized form as appropriate – although it is desirable for the database identifier(s) to reference to the biological (non-derivatized) form. In general, different adduct forms would generally be reported in the Small Molecule Feature section.
The order of columns MUST follow the order specified below.
All columns are MANDATORY except for “opt_” columns.
6.3.1. SML_ID
Description |
A within file unique identifier for the small molecule. |
Type |
Integer |
Is Nullable: |
FALSE |
Example |
SMH SML_ID … SML 1 … SML 2 … |
6.3.2. SMF_ID_REFS
Description |
References to all the features on which quantitation has been based (SMF elements) via referencing SMF_ID values. Multiple values SHOULD be provided as a “|” separated list. This MAY be null only if this is a Summary file. |
Type |
{SMF_ID} list |
Is Nullable: |
TRUE |
Example |
SMH SML_ID SMF_ID_REFS SML 1 2|3|11… |
6.3.3. database_identifier
Description |
A list of “|” separated possible identifiers for the small molecule; multiple values MUST only be provided to indicate ambiguity in the identification of the molecule and not to demonstrate different identifier types for the same molecule. Alternative identifiers for the same molecule MAY be provided as optional columns. The database identifier must be preceded by the resource description (prefix) followed by a colon, as specified in the metadata section. A null value MAY be provided if the identification is sufficiently ambiguous as to be meaningless for reporting or the small molecule has not been identified. |
Type |
String List |
Is Nullable: |
TRUE |
Example |
SMH SML_ID database_identifier … SML 1 CID:00027395 … SML 2 HMDB:HMDB0001847 SML 3 null |
6.3.4. chemical_formula
Description |
A list of “|” separated potential chemical formulae of the reported compound. The number of values provided MUST match the number of entities reported under “database_identifier”, even if this leads to redundant reporting of information (i.e. if ambiguity can be resolved in the chemical formula), and the validation software will throw an error if the number of “|” symbols does not match. “null” values between bars are allowed. This should be specified in Hill notation (EA Hill 1900), i.e. elements in the order C, H and then alphabetically all other elements. Counts of one may be omitted. Elements should be capitalized properly to avoid confusion (e.g., “CO” vs. “Co”). The chemical formula reported should refer to the neutral form. Example N-acetylglucosamine would be encoded by the string “C8H15NO6” |
Type |
String List |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … chemical_formula … SML 1 … C17H20N4O2 … |
6.3.5. smiles
Description |
A list of “|” separated potential molecule structures in the simplified molecular-input line-entry system (SMILES) for the small molecule. The number of values provided MUST match the number of entities reported under “database_identifier”, and the validation software will throw an error if the number of “|” symbols does not match. “null” values between bars are allowed. |
Type |
String List |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … chemical_formula smiles … SML 1 … C17H20N4O2 C1=CC=C(C=C1)CCNC(=O)CCNNC(=O)C2=CC=NC=C2 … |
6.3.6. inchi
Description |
A list of “|” separated potential standard IUPAC International Chemical Identifier (InChI) of the given substance. The number of values provided MUST match the number of entities reported under “database_identifier”, even if this leads to redundant information being reported (i.e. if ambiguity can be resolved in the InChi), and the validation software will throw an error if the number of “|” symbols does not match. “null” values between bars are allowed. |
Type |
String List |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … chemical_formula … inchi … SML 1 … C17H20N4O2 … InChI=1S/C17H20N4O2/c22-16(19-12-6-14-4-2-1-3-5-14)9-13-20-21-17(23)15-7-10-18-11-8-15/h1-5,7-8,10-11,20H,6,9,12-13H2,(H,19,22)(H,21,23) … |
6.3.7. chemical_name
Description |
A list of “|” separated possible chemical/common names for the small molecule, or general description if a chemical name is unavailable. Multiple names are only to demonstrate ambiguity in the identification. The number of values provided MUST match the number of entities reported under “database_identifier”, and the validation software will throw an error if the number of “|” symbols does not match. “null” values between bars are allowed. |
Type |
String List |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … description … SML 1 … N-(2-phenylethyl)-3-[2-(pyridine-4-carbonyl)hydrazinyl]propanamide… |
6.3.8. uri
Description |
A URI pointing to the small molecule’s entry in a reference database (e.g., the small molecule’s HMDB or KEGG entry). The number of values provided MUST match the number of entities reported under “database_identifier”, and the validation software will throw an error if the number of “|” symbols does not match. “null” values between bars are allowed. |
Type |
URI List |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … uri … SML 1 … http://www.genome.jp/dbget-bin/www_bget?cpd:C00031 … SML 2 … http://www.hmdb.ca/metabolites/HMDB0001847 … SML 3 … http://identifiers.org/hmdb/HMDB0001847 … |
6.3.9. theoretical_neutral_mass
Description |
The small molecule’s precursor’s theoretical neutral mass. The number of values provided MUST match the number of entities reported under “database_identifier”, and the validation software will throw an error if the number of “|” symbols does not match. “null” values (in general and between bars) are allowed for molecules that have not been identified only, or for molecules where the neutral mass cannot be calculated. In these cases, the SML entry SHOULD reference features in which exp_mass_to_charge values are captured. |
Type |
Double List |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … theoretical_neutral_mass … SML 1 … 1234.5 … |
6.3.10. adduct_ions
Description |
A “|” separated list of detected adducts for this this molecule, following the general style in the 2013 IUPAC recommendations on terms relating to MS e.g. |
Type |
Regex List \[\d*M([+-][\w\d]+)*\]\d*[+-] |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … adduct_ions … SML 1 … [M+H]1+ | [M+Na]1+ … |
6.3.11. reliability
Description |
The reliability of the given small molecule identification. This must be supplied by the resource and MUST be reported as an integer between 1-4:
These MAY be replaced using a suitable CV term in the metadata section e.g. to use MSI recommendation levels (see Section 6.2.57 for details). The following CV terms are already available within the PSI MS CV. Future schemes may be implemented by extending the PSI MS CV with new terms and associated levels. The MSI has recently discussed an extension of the original four level scheme into a five level scheme MS:1002896 (compound identification confidence level) with levels
For high-resolution MS, the following term and its levels may be used: MS:1002955 (hr-ms compound identification confidence level) with levels
A String data type is set to allow for different systems to be specified in the metadata section. |
Type |
String |
Is Nullable: |
TRUE |
Example |
SMH identifier … reliability … SML 1 … 3 … or MTD small_molecule-identification_reliability [MS, MS:1002896, compound identification confidence level,] … SMH identifier … reliability … SML 1 … 0 … or MTD small_molecule-identification_reliability [MS, MS:1002955, hr-ms compound identification confidence level,] … SMH identifier … reliability … SML 1 … 2a … |
6.3.12. best_id_confidence_measure
Description |
The approach or database search that identified this small molecule with highest confidence. |
Type |
Parameter |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … best_ id_confidence_measure … SML 1 … [MS, MS:1001477, SpectraST,] … |
6.3.13. best_id_confidence_value
Description |
The best confidence measure in identification (for this type of score) for the given small molecule across all assays. The type of score MUST be defined in the metadata section. If the small molecule was not identified by the specified search engine, “null” MUST be reported. If the confidence measure does not report a numerical confidence value, “null” SHOULD be reported. |
Type |
Double |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … best_id_confidence_value … SML 1 … 0.7 … |
6.3.14. abundance_assay[1-n]
Description |
The small molecule’s abundance in every assay described in the metadata section MUST be reported. Null or zero values may be reported as appropriate. "null" SHOULD be used to report missing quantities, while zero SHOULD be used to indicate a present but not reliably quantifiable value (e.g. below a minimum noise threshold). |
Type |
Double |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … abundance_assay[1] … SML 1 … 0.3 … |
6.3.15. abundance_study_variable[1-n]
Description |
The small molecule’s abundance in all the study variables described in the metadata section (study_variable[1-n]_average_function), calculated using the method as described in the Metadata section (default = arithmetic mean across assays). Null or zero values may be reported as appropriate. "null" SHOULD be used to report missing quantities, while zero SHOULD be used to indicate a present but not reliably quantifiable value (e.g. below a minimum noise threshold). |
Type |
Double |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … abundance_study_variable[1] … SML 1 … 0.3 … |
6.3.16. abundance_variation_study_variable [1-n]
Description |
A measure of the variability of the study variable abundance measurement, calculated using the method as described in the metadata section (study_variable[1-n]_average_function), with a default = arithmethic co-efficient of variation of the small molecule’s abundance in the given study variable. |
Type |
Double |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … abundance_study_variable[1] abundance_variation_study_variable[1]… SML 1 … 0.3 0.04 … |
6.3.17. opt_{identifier}_*
Description |
Additional columns can be added to the end of the small molecule table. These column headers MUST start with the prefix “opt_” followed by the {identifier} of the object they reference: assay, study variable, MS run or “global” (if the value relates to all replicates). Column names MUST only contain the following characters: ‘A’-‘Z’, ‘a’-‘z’, ‘0’-‘9’, ‘’, ‘-’, ‘[’, ‘]’, and ‘:’. CV parameter accessions MAY be used for optional columns following the format: opt_{identifier}_cv_{accession}_{parameter name}. Spaces within the parameter’s name MUST be replaced by ‘’. |
Type |
Column |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … opt_assay[1]_my_value … opt_global_another_value SML 1 … My value … some other value |
Example optional columns:
-
Species
-
Taxid
-
GO term IDs
-
Retention time index values normalised to a given scale
-
Identification scores specific to each assay
-
Raw quantification values, assuming normalised values are provided in the standard assay quantification columns.
6.4. Small Molecule Feature (SMF) Section
The small molecule feature section is table-based, representing individual MS regions (generally considered to be the elution profile for all isotopomers formed from a single charge state of a molecule), that have been measured/quantified. However, for approaches that quantify individual isotopomers e.g. stable isotope labelling/flux studies, then each SMF row SHOULD represent a single isotopomer.
Different adducts or derivatives and different charge states of individual molecules should be reported as separate SMF rows.
The small molecule feature section MUST always come after the Small Molecule Table. All table columns MUST be Tab separated. There MUST NOT be any empty cells. Missing values MUST be reported using “null”.
The order of columns MUST follow the order specified below.
All columns are MANDATORY except for “opt_” columns.
6.4.1. SMF_ID
Description |
A within file unique identifier for the small molecule feature. |
Type |
Integer |
Is Nullable: |
FALSE |
Example |
SFH SMF_ID … SMF 1 … SMF 2 … |
6.4.2. SME_ID_REFS
Description |
References to the identification evidence (SME elements) via referencing SME_ID values. Multiple values MAY be provided as a “|” separated list to indicate ambiguity in the identification or to indicate that different types of data supported the identifiction (see SME_ID_REF_ambiguity_code). For the case of a consensus approach where multiple adduct forms are used to infer the SML ID, different features should just reference the same SME_ID value(s). |
Type |
{SME_ID} list |
Is Nullable: |
TRUE |
Example |
SFH SMF_ID SME_ID_REFS SMF 1 5|6|12… |
6.4.3. SME_ID_REF_ambiguity_code
Description |
If multiple values are given under SME_ID_REFS, one of the following codes MUST be provided. 1=Ambiguous identification; 2=Only different evidence streams for the same molecule with no ambiguity; 3=Both ambiguous identification and multiple evidence streams. If there are no or one value under SME_ID_REFs, this MUST be reported as null. |
Type |
Integer |
Is Nullable: |
TRUE |
Example |
SFH SMF_ID SME_ID_REFS SME_ID_REF_ambiguity_code SMF 1 5|6|12… 1 |
6.4.4. adduct_ion
Description |
The assumed classification of this molecule’s adduct ion after detection, following the general style in the 2013 IUPAC recommendations on terms relating to MS e.g. |
Type |
Regex \[\d*M([+-][\w\d]+)*\]\d*[+-] |
Is Nullable: |
TRUE |
Example |
SFH SMF_ID … adduct_ion … SMF 1 … [M+H]+ … SMF 2 … [M+2Na]2+ … |
6.4.5. isotopomer
Description |
If de-isotoping has not been performed, then the isotopomer quantified MUST be reported here e.g. “+1”, “+2”, “13C peak” using CV terms, otherwise (i.e. for approaches where SMF rows are de-isotoped features) this MUST be null. |
Type |
Parameter |
Is Nullable: |
TRUE |
Example |
SFH SMF_ID … isotopomer … SMF 1 … [MS,MS:1002957,”isotopomer MS peak”,”13C peak”]… |
6.4.6. exp_mass_to_charge
Description |
The experimental mass/charge value for the feature, by default assumed to be the mean across assays or a representative value. For approaches that report isotopomers as SMF rows, then the m/z of the isotopomer MUST be reported here. |
Type |
Double |
Is Nullable: |
FALSE |
Example |
SFH SMF_ID … exp_mass_to_charge … SMF 1 … 1234.5 … |
6.4.7. charge
Description |
The feature’s charge value using positive integers both for positive and negative polarity modes. |
Type |
Integer |
Is Nullable: |
FALSE |
Example |
SFH SMF_ID … charge … SMF 1 … 1 … |
6.4.8. retention_time_in_seconds
Description |
The apex of the feature on the retention time axis, in a Master or aggregate MS run. Retention time MUST be reported in seconds. Retention time values for individual MS runs (i.e. before alignment) MAY be reported as optional columns. Retention time SHOULD only be null in the case of direct infusion MS or other techniques where a retention time value is absent or unknown. Relative retention time or retention time index values MAY be reported as optional columns, and could be considered for inclusion in future versions of mzTab as appropriate. |
Type |
Double |
Is Nullable: |
TRUE |
Example |
SFH SMF_ID … retention_time_in_seconds … SMF 1 … 1345.7 … |
6.4.9. retention_time_in_seconds_start
Description |
The start time of the feature on the retention time axis, in a Master or aggregate MS run. Retention time MUST be reported in seconds. Retention time start and end SHOULD only be null in the case of direct infusion MS or other techniques where a retention time value is absent or unknown and MAY be reported in optional columns. |
Type |
Double |
Is Nullable: |
TRUE |
Example |
SFH SMF_ID … retention_time_in_seconds_start … SMF 1 … 1327.0 … |
6.4.10. retention_time_in_seconds_end
Description |
The end time of the feature on the retention time axis, in a Master or aggregate MS run. Retention time MUST be reported in seconds. Retention time start and end SHOULD only be null in the case of direct infusion MS or other techniques where a retention time value is absent or unknown and MAY be reported in optional columns.. |
Type |
Double |
Is Nullable: |
TRUE |
Example |
SFH SMF_ID … retention_time_in_seconds_end … SMF 1 … 1327.8 … |
6.4.11. abundance_assay[1-n]
Description |
The feature’s abundance in every assay described in the metadata section MUST be reported. Null or zero values may be reported as appropriate. |
Type |
Double |
Is Nullable: |
TRUE |
Example |
SMH SML_ID … abundance_assay[1] … SMF 1 … 38648 … |
6.4.12. opt_{identifier}_*
Description |
Additional columns can be added to the end of the small molecule feature table. These column headers MUST start with the prefix “opt_” followed by the {identifier} of the object they reference: assay, study variable, MS run or “global” (if the value relates to all replicates). Column names MUST only contain the following characters: ‘A’-‘Z’, ‘a’-‘z’, ‘0’-‘9’, ‘’, ‘-’, ‘[’, ‘]’, and ‘:’. CV parameter accessions MAY be used for optional columns following the format: opt_{identifier}_cv_{accession}_{parameter name}. Spaces within the parameter’s name MUST be replaced by ‘’. |
Type |
Column |
Is Nullable: |
TRUE |
Example |
SFH SMF_ID … opt_assay[1]_my_value … opt_global_another_value SMF 1 … My value … some other value |
Example optional columns:
-
(Apex) retention time values for each MS run pre-alignment
-
Retention time index values normalised to a given scale
-
Raw quantification values, assuming normalised values are provided in the standard assay quantification columns.
-
Predicted retention time
-
CCS values
-
Two- or n-dimensional retention times e.g.
opt_global_retention_time_ndopt_global_retention_time_nd_window_startopt_global_retention_time_nd_window_end
6.5. Small Molecule Evidence (SME) Section
The small molecule evidence section is table-based, representing evidence for identifications of small molecules/features, from database search or any other process used to give putative identifications to molecules. In a typical case, each row represents one result from a single search or intepretation of a piece of evidence e.g. a database search with a fragmentation spectrum. Multiple results from a given input data item (e.g. one fragment spectrum) SHOULD share the same value under evidence_input_id.
The small molecule evidence section MUST always come after the Small Molecule Feature Table. All table columns MUST be Tab separated. There MUST NOT be any empty cells. Missing values MUST be reported using “null”.
The order of columns MUST follow the order specified below.
All columns are MANDATORY except for “opt_” columns.
6.5.1. SME_ID
Description |
A within file unique identifier for the small molecule evidence result. |
Type |
Integer |
Is Nullable: |
FALSE |
Example |
SEH SME_ID … SME 1 … |
6.5.2. evidence_input_id
Description |
A within file unique identifier for the input data used to support this identification e.g. fragment spectrum, RT and m/z pair, isotope profile that was used for the identification process, to serve as a grouping mechanism, whereby multiple rows of results from the same input data share the same ID. The identifiers may be human readable but should not be assumed to be interpretable. For example, if fragmentation spectra have been searched then the ID may be the spectrum reference, or for accurate mass search, the ms_run[2]:458.75. |
Type |
String |
Is Nullable: |
FALSE |
Example |
SEH SME_ID evidence_input_id … SME 1 ms_run[1]:mass=278.65;rt=376.5 SME 2 ms_run[1]:mass=278.65;rt=376.5 SME 3 ms_run[1]:mass=278.65;rt=376.5 (in this example three identifications were made from the same accurate mass/RT library search) |
6.5.3. database_identifier
Description |
The putative identification for the small molecule sourced from an external database, using the same prefix specified in database[1-n]-prefix. This could include additionally a chemical class or an identifier to a spectral library entity, even if its actual identity is unknown. For the “no database” case, "null" must be used. The unprefixed use of "null" is prohibited for any other case. If no putative identification can be reported for a particular database, it MUST be reported as the database prefix followed by null. |
Type |
String |
Is Nullable: |
TRUE |
Example |
SEH SME_ID identifier … SME 1 CID:00027395 … SME 2 HMDB:HMDB12345 … SME 3 CID:null … |
6.5.4. chemical_formula
Description |
The chemical formula of the identified compound e.g. in a database, assumed to match the theoretical mass to charge (in some cases this will be the derivatized form, including adducts and protons). This should be specified in Hill notation (EA Hill 1900), i.e. elements in the order C, H and then alphabetically all other elements. Counts of one may be omitted. Elements should be capitalized properly to avoid confusion (e.g., “CO” vs. “Co”). The chemical formula reported should refer to the neutral form. Charge state is reported by the charge field. Example N-acetylglucosamine would be encoded by the string “C8H15NO6” |
Type |
String |
Is Nullable: |
TRUE |
Example |
SEH SME_ID … chemical_formula … SME 1 … C17H20N4O2 … |
6.5.5. smiles
Description |
The potential molecule’s structure in the simplified molecular-input line-entry system (SMILES) for the small molecule. |
Type |
String |
Is Nullable: |
TRUE |
Example |
SEH SME_ID … chemical_formula smiles … SML 1 … C17H20N4O2 C1=CC=C(C=C1)CCNC(=O)CCNNC(=O)C2=CC=NC=C2 … |
6.5.6. inchi
Description |
A standard IUPAC International Chemical Identifier (InChI) for the given substance. |
Type |
String |
Is Nullable: |
TRUE |
Example |
SEH SME_ID … chemical_formula … inchi … SML 1 … C17H20N4O2 … InChI=1S/C17H20N4O2/c22-16(19-12-6-14-4-2-1-3-5-14)9-13-20-21-17(23)15-7-10-18-11-8-15/h1-5,7-8,10-11,20H,6,9,12-13H2,(H,19,22)(H,21,23) … |
6.5.7. chemical_name
Description |
The small molecule’s chemical/common name, or general description if a chemical name is unavailable. |
Type |
String |
Is Nullable: |
TRUE |
Example |
SEH SME_ID … chemical_name … SML 1 … N-(2-phenylethyl)-3-[2-(pyridine-4-carbonyl)hydrazinyl]propanamide… |
6.5.8. uri
Description |
A URI pointing to the small molecule’s entry in a database (e.g., the small molecule’s HMDB, Chebi or KEGG entry). |
Type |
URI |
Is Nullable: |
TRUE |
Example |
SEH SME_ID … uri … SME 1 … http://www.hmdb.ca/metabolites/HMDB00054 |
6.5.9. derivatized_form
Description |
If a derivatized form has been analysed by MS, then the functional group attached to the molecule should be reported here using suitable userParam or CV terms as appropriate. |
Type |
Parameter |
Is Nullable: |
TRUE |
Example |
COM This example shows a triple substitution with a TMS group (3TMS) SMH database_identifier … derivatized_form … SML CID:00027395 … [CHEBI, CHEBI:51088, trimethylsilyl group, 3] … |
6.5.10. adduct_ion
Description |
The assumed classification of this molecule’s adduct ion after detection, following the general style in the 2013 IUPAC recommendations on terms relating to MS e.g. |
Type |
Regex \[\d*M([+-][\w\d]+)*\]\d*[+-] |
Is Nullable: |
TRUE |
Example |
SEH SME_ID … adduct_ion … SME 1 … [M+H]+ … SME 2 … [M+2Na]2+ … OR (for negative mode): SME 1 … [M-H]- … SME 2 … [M+Cl]- … |
6.5.11. exp_mass_to_charge
Description |
The experimental mass/charge value for the precursor ion. If multiple adduct forms have been combined into a single identification event/search, then a single value e.g. for the protonated form SHOULD be reported here. |
Type |
Double |
Is Nullable: |
FALSE |
Example |
SEH SME_ID … exp_mass_to_charge … SME 1 … 1234.5 … |
6.5.12. charge
Description |
The small molecule evidence’s charge value using positive integers both for positive and negative polarity modes. |
Type |
Integer |
Is Nullable: |
FALSE |
Example |
SEH SME_ID … charge … SME 1 … 1 … |
6.5.13. theoretical_mass_to_charge
Description |
The theoretical mass/charge value for the small molecule or the database mass/charge value (for a spectral library match). |
Type |
Double |
Is Nullable: |
FALSE |
Example |
SEH SME_ID … theoretical_mass_to_charge … SME 1 … 1234.71 … |
6.5.14. spectra_ref
Description |
Reference to a spectrum in a spectrum file, for example a fragmentation spectrum has been used to support the identification. If a separate spectrum file has been used for fragmentation spectrum, this MUST be reported in the metadata section as additional ms_runs. The reference must be in the format ms_run[1-n]:{SPECTRA_REF} where SPECTRA_REF MUST follow the format defined in 5.2 (including references to chromatograms where these are used to inform identification). Multiple spectra MUST be referenced using a “|” delimited list for the (rare) cases in which search engines have combined or aggregated multiple spectra in advance of the search to make identifications. If a fragmentation spectrum has not been used, the value should indicate the ms_run to which is identification is mapped e.g. “ms_run[1]”. |
Type |
String List |
Is Nullable: |
FALSE |
Example |
SEH SME_ID … spectra_ref … SME 1 … ms_run[1]:index=5 … |
6.5.15. identification_method
Description |
The database search, search engine or process that was used to identify this small molecule e.g. the name of software, database or manual curation etc. If manual validation has been performed quality, the following CV term SHOULD be used: "quality estimation by manual validation" MS:1001058. |
Type |
Parameter |
Is Nullable: |
FALSE |
Example |
SEH SME_ID … identification_method… SME 1 … [MS, MS:1001477, SpectraST,] … |
6.5.16. ms_level
Description |
The highest MS level used to inform identification e.g. MS1 (accurate mass only) = “ms level=1” or from an MS2 fragmentation spectrum = “ms level=2”. For direct fragmentation or data independent approaches where fragmentation data is used, appropriate CV terms SHOULD be used . |
Type |
Parameter |
Is Nullable: |
FALSE |
Example |
SEH SME_ID … ms_level … SME 1 … [MS, MS:1000511, ms level, 2] … |
6.5.17. id_confidence_measure[1-n]
Description |
Any statistical value or score for the identification. The metadata section reports the type of score used, as id_confidence_measure[1-n] of type Param. |
Type |
Double |
Is Nullable: |
TRUE |
Example |
MTD id_confidence_measure[1] [MS, MS:1001419, SpectraST:discriminant score F,] … SEH SME_ID … id_confidence_measure[1] … SME 1 … 0.7 … |
6.5.18. rank
Description |
The rank of this identification from this approach as increasing integers from 1 (best ranked identification). Ties (equal score) are represented by using the same rank – defaults to 1 if there is no ranking system used. |
Type |
Integer |
Is Nullable: |
FALSE |
Example |
SEH SME_ID … rank … SME 1 … 1 … |
6.5.19. opt_{identifier}_*
Description |
Additional columns can be added to the end of the small molecule evidence table. These column headers MUST start with the prefix “opt_” followed by the {identifier} of the object they reference: assay, study variable, MS run or “global” (if the value relates to all replicates). Column names MUST only contain the following characters: ‘A’-‘Z’, ‘a’-‘z’, ‘0’-‘9’, ‘’, ‘-’, ‘[’, ‘]’, and ‘:’. CV parameter accessions MAY be used for optional columns following the format: opt_{identifier}_cv_{accession}_{parameter name}. Spaces within the parameter’s name MUST be replaced by ‘’. |
Type |
Column |
Is Nullable: |
TRUE |
Example |
SEH SME_ID … opt_assay[1]_my_value … opt_global_another_value SML 1 … My value … some other value |
Example optional columns:
-
Additional statistical measures or annotations about evidence, such as decoy identifications or rules used for fragment-based identification.
7. Non-supported use cases
There are a number of use cases that were discussed during the development process and it was decided that they are not explicitly supported in mzTab version 2.0.0-M. They may be implemented in future versions of the standard.
Examples include:
-
Multiplexing technologies
-
Including the results from different technologies in one mzTab file e.g. DIMS and LC/MS
-
Merging of results from different omics experiments, e.g. proteomics, metabolomics and lipidomics
8. Conclusions
This document contains the specifications for using the mzTab format to represent results from small molecule pipelines, in the context of a metabolomics or lipidomics investigation. This specification constitutes a proposal for a standard from the Proteomics Standards Initiative and Metabolomics Standards Initiative. These artefacts are currently undergoing the PSI document process, which will result in a standard officially sanctioned by PSI/MSI.
9. Reference Implementation
A reference implementation in JAVA is available at https://github.com/lifs-tools/jmzTab-m. The reference implementation provides a parser, a validator, a CV-mapping validation and a writer for mzTab-M. It furthermore supports transcoding from a JSON representation of the object model into the tab-separated output format and vice-versa. A user-friendly web-application that uses the validator reference implementation is available at https://apps.lifs.isas.de/mztabvalidator/.
10. Authors
-
Nils Hoffmann, Leibniz-Institut für Analytische Wissenschaften – ISAS – e.V., Dortmund, Germany. nils.hoffmann@isas.de
-
Joel Rein, Wellcome Sanger Institute, Cambridge, United Kingdom. joel.rein@sanger.ac.uk
-
Timo Sachsenberg, Applied Bioinformatics Group, Center for Bioinformatics, University of Tübingen, Germany. sachsenb@informatik.uni-tuebingen.de
-
Jürgen Hartler, Institute of Computational Biotechnology at Graz University of Technology and Center for Explorative Lipidomics, Graz, Austria. juergen.hartler@tugraz.at
-
Kenneth Haug, European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Cambridge, United Kingdom. kenneth@ebi.ac.uk
-
Gerhard Mayer, Medizinisches Proteom-Center, Ruhr-Universität Bochum, Germany. gerhard.mayer@rub.de
-
Oliver Alka, Applied Bioinformatics Group, Center for Bioinformatics, University of Tübingen, Germany. alka@informatik.uni-tuebingen.de
-
Saravanan Dayalan, Metabolomics Australia, The University of Melbourne, Parkville, Australia. sdayalan@unimelb.edu.au
-
Jake TM Pearce, MRC-NIHR National Phenome Center, Imperial College London, London, United Kingdom. jake.pearce@imperial.ac.uk
-
Philippe Rocca-Serra, Oxford e-Research Centre, University of Oxford, United Kingdom. philippe.rocca-serra@oerc.ox.ac.uk
-
Da Qi, Institute of Integrative Biology, University of Liverpool, United Kingdom and BGI-Shenzhen, Shenzen, China. qida@genomics.cn
-
Martin Eisenacher, Medizinisches Proteom-Center, Ruhr-Universität Bochum, Germany. martin.eisenacher@ruhr-uni-bochum.de
-
Yasset Perez-Riverol, European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Cambridge, United Kingdom. yperez@ebi.ac.uk
-
Juan Antonio Vizcaíno, European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Cambridge, United Kingdom. juan@ebi.ac.uk
-
Reza M Salek, International Agency for Research on Cancer, Lyon, France. r7salek@gmail.com
-
Steffen Neumann, Leibniz Institute of Plant Biochemistry, Halle and German Centre for Integrative Biodiversity Researchm Halle-Jena-Leipzig, Germany. sneumann@ipb-halle.de
-
Andrew R Jones, Institute of Integrative Biology, University of Liverpool, United Kingdom. (Editor) Andrew.Jones@liverpool.ac.uk
References
-
[bradner-1997] Bradner, S. (1997). Key words for use in RFCs to Indicate Requirement Levels, Internet Engineering Task Force. RFC 2119.
-
[martens-2011] Martens, L., et al. (2011). "mzML—a community standard for mass spectrometry data." Mol Cell Proteomics 10(1): R110 000133.
-
[hill-1900] EA Hill (1900). “ON A SYSTEM OF INDEXING CHEMICAL LITERATURE; ADOPTED BY THE CLASSIFICATION DIVISION OF THE U. S. PATENT OFFICE.” J. Am. Chem. Soc. 22 (8): 478–494. doi:10.1021/ja02046a005.
-
[griss-2014] Griss et al. (2014) "The mzTab data exchange format: communicating mass-spectrometry-based proteomics and metabolomics experimental results to a wider audience." Mol Cell Proteomics doi: 10.1074/mcp.O113.036681.
-
[sansone-2012] Sansone et al. (2012) "Toward interoperable bioscience data." Nature Genetics 44: 121–126. doi:10.1038/ng.1054.
11. Intellectual Property Statement
The PSI/MSI takes no position regarding the validity or scope of any intellectual property or other rights that might be claimed to pertain to the implementation or use of the technology described in this document or the extent to which any license under such rights might or might not be available; neither does it represent that it has made any effort to identify any such rights. Copies of claims of rights made available for publication and any assurances of licenses to be made available, or the result of an attempt made to obtain a general license or permission for the use of such proprietary rights by implementers or users of this specification can be obtained from the PSI Chair.
The PSI/MSI invites any interested party to bring to its attention any copyrights, patents or patent applications, or other proprietary rights that may cover technology that may be required to practice this recommendation. Please address the information to the PSI Chair (see contacts information at PSI website).
TradeMark Section
Microsoft Excel®
Copyright Notice
Copyright © Proteomics Standards Initiative (2018). All Rights Reserved.
This document and translations of it may be copied and furnished to others, and derivative works that comment on or otherwise explain it or assist in its implementation may be prepared, copied, published and distributed, in whole or in part, without restriction of any kind, provided that the above copyright notice and this paragraph are included on all such copies and derivative works. However, this document itself may not be modified in any way, such as by removing the copyright notice or references to the PSI or other organizations, except as needed for the purpose of developing Proteomics Recommendations in which case the procedures for copyrights defined in the PSI Document process must be followed, or as required to translate it into languages other than English.
The limited permissions granted above are perpetual and will not be revoked by the PSI or its successors or assigns.
This document and the information contained herein is provided on an "AS IS" basis and THE PROTEOMICS STANDARDS INITIATIVE DISCLAIMS ALL WARRANTIES, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO ANY WARRANTY THAT THE USE OF THE INFORMATION HEREIN WILL NOT INFRINGE ANY RIGHTS OR ANY IMPLIED WARRANTIES OF MERCHANTABILITY OR FITNESS FOR A PARTICULAR PURPOSE."